
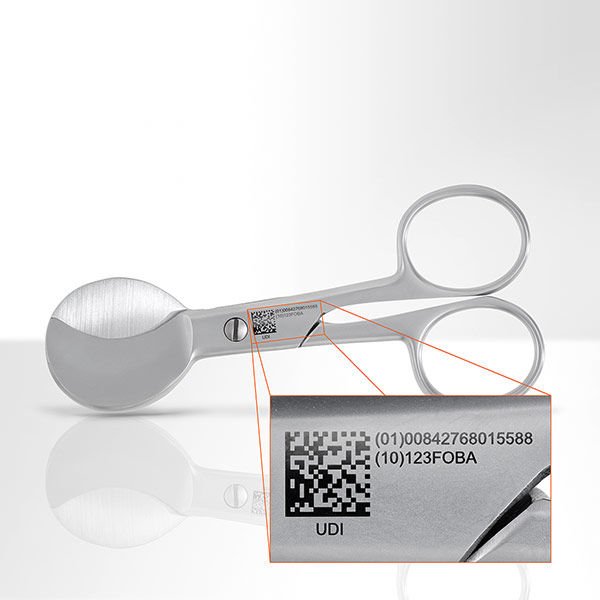
Die Idee ist grundsätzlich lobenswert, denn es geht um die Sicherheit der Patienten und die Rückverfolgbarkeit von Medizinprodukten – weltweit: Zukünftig soll jedes Medizinprodukt eindeutig gekennzeichnet und damit identifizierbar sein. Dazu erhält es eine Produktidentifikation, im Englischen „Unique Device Identification“, kurz „UDI“. Diese UDI soll in Form eines Zahlencodes von Menschenaugen und in Form, zum Beispiel eines Strich- oder 2D-Codes, von Maschinen lesbar sein. So wurde es im Auftrag der US-amerikanischen Food and Drug Administration (FDA) diskutiert und auf internationaler Ebene beschlossen. Auf europäischer Ebene lautet die dazugehörige Verordnung „Medical Device Regulation (MDR)“. Sie wurde bereits 2017 eingeführt.
Inhaltsverzeichnis
1. UDI und MDR: Ende der Übergangszeit naht
2. Verschiebung für Eudamed
3. Neuzertifizierung von Medizinprodukten gemäß MDR
4. Benannte Stellen sind der Knackpunkt
5. Zertifikate nach MDD als Ausweg
6. MDR und UDI: Abwarten ist keine Lösung
7. UDI-konform, MDR-konform: Keine Kleinigkeit
8. Aufwand ist für KMU kaum zu stemmen
9. Basiswissen Unique Device Identification (UDI)
10. Sonderrolle Basic UDI-DI
11. Wo muss die UDI erscheinen?
12. Ab wann UDI?
UDI und MDR: Ende der Übergangszeit naht
Dass die Diskussionen um die MDR und in deren Zusammenhang um die UDI nun erst so richtig hochkochen, liegt an der eingeräumten Übergangszeit, die bald ausläuft – und noch einigen Unklarheiten. Nichtsdestotrotz gilt‘s ab dem 26. Mai 2020, und Medizinproduktehersteller, die ihre Produkte innerhalb der EU vertreiben wollen, müssen diesen eine eindeutige UDI zuweisen. Mit den dazugehörigen Stammdaten und einer eindeutigen so genannten Basis-UDI soll diese innerhalb einer Übergangsfrist von (zurzeit) 18 Monaten an die UDI-Datenbank der EU gemeldet werden, die ein Teil der EU-Datenbank Eudamed sein wird.
Wie das grundsätzlich aussehen kann, hat die FDA bereits vorgemacht: Sie hat die UDI schon verpflichtend eingeführt. Gesammelt werden hier die Daten in der US-amerikanischen „Global Unique Device Identification Database“ (GUDID). Man könnte meinen, dass, wer das Prozedere für den amerikanischen Markt hinter sich hat, auch für die anstehende UDI-Einführung auf EU-Ebene recht gut gerüstet sei.
Verschiebung für Eudamed
Die Einführung der Datenbank wurde jedoch verschoben. Ursprünglich sollte die zentrale Datenbank ab Mai 2020 zur Verfügung stehen. Die EU-Kommission hat diesen Termin jedoch im Herbst 2019 nochmal um zwei Jahre nach hinten verlegt.
Neuzertifizierung von Medizinprodukten gemäß MDR
Durch die Erfahrungen in den USA gut gerüstet? Eigentlich, das Ganze hat aber leider einen Haken: Die „FDA-UDI“ lässt sich nicht 1:1 auf die „EU-UDI“ übertragen. Es gibt kein weltweit einheitliches Nummernsystem. Das wäre zu schön und einfach. Und apropos einfach: Die Voraussetzungen für die „FDA-UDI“ sind weitaus einfacher, denn die UDI-Datenbank der FDA ist nicht direkt mit dem Zulassungsprozedere verknüpft.
Die EU hingegen hat mit der MDR gleich den ganz großen Wurf versucht. Hier hängt die Zertifizierungs- mit der UDI-Datenbank zusammen. Damit ist der ganze Prozess der durch die MDR geforderten Neuzertifizierung von Medizinprodukten eng mit der UDI verknüpft.
Und hier liegt das eigentliche Problem: Für diese Neuzertifizierung gibt es bisher, Stand Mitte Oktober 2019, insgesamt nur fünf benannte Stellen – gegenüber 58 nach alter Medizinprodukterichtlinie (MDD): in Deutschland sind dies die Dekra, der TÜV Rheinland und der TÜV Süd.
Benannte Stellen sind der Knackpunkt
Weitere Stellen sollen folgen, stecken aber noch im Benennungsprozess. Bis sie voll einsatzbereit sind, kann es noch dauern, denn: „Das Regelsystem der MDR ist noch in ständiger Veränderung“, weiß Dr. Bassil Akra, Leiter strategische Geschäftsentwicklung der Medizinproduktesparte, TÜV Süd, aus eigener Erfahrung. „Wir waren eine der ersten zwei benannten Stellen für die MDR und haben erst zwei Zertifikate erstellt, weil es immer wieder Neuinterpretationen auf EU-Ebene gibt. Wir stecken in einer Art ‚Kalibrierungsphase‘, die sich wahrscheinlich bis Ende 2019 hinziehen wird.“
Und das, obwohl sich der Anteil der von den Benannten Stellen zu zertifizierenden Produkte durch die neue MDR erhöht. Eine allein von den Herstellern ausgefüllte Konformitätserklärung reicht zum Beispiel für wiederverwendbare chirurgische Instrumente und medizinische Software nicht mehr aus.
Zertifikate nach MDD als Ausweg
Um mehr Zeit zu bekommen, erneuern zurzeit viele Unternehmen die Zertifikate ihre Produkte nach der noch geltenden Medical Device Directive (MDD), denn diese gelten dann noch über den 26. Mai 2020 hinaus. Das Ganze hat aber einen Haken: Laut MDR müssen auch diese Produkte ab Mai 2020 Anforderungen der MDR erfüllen – zum Beispiel die Überwachung nach dem Inverkehrbringen und die Marktüberwachung.
Die Verunsicherung bei den Herstellern ist daher groß. Meinrad Kempf, Projektmanager bei der MedicalMountains GmbH in Tuttlingen, berichtet: „Viele Unternehmen hätten gerne schon mit der Neuzertifizierung eines Produktes als ‚Testballon‘ begonnen, um zu erfahren, was eventuell noch fehlen könnte. Doch die benannten Stellen sind noch nicht soweit. Daher muss jetzt der erste Versuch gleich passen.“
MDR und UDI: Abwarten ist keine Lösung
Abwarten ist jedoch auch keine Lösung, warnt hingegen Dr. Bassil Akra vom TÜV Süd: „Die Unternehmen müssen mit der von ihnen gewählten benannten Stelle zwei Dinge klären. Erstens, wann und ob diese für die neue MDR benannt wird, und zweitens, ob sie dann noch alle Produkte des Herstellers nach der MDR zertifizieren darf – da ändert sich einiges. Es wird voraussichtlich nur wenige benannte Stellen geben, die Produkte der Klasse III zertifizieren können. Wir sind eine davon.“
Ein weiterer Knackpunkt, der vielen Unternehmen noch nicht klar sei: „Für die Zertifizierung müssen alle Anforderungen erfüllt sein, es kann nichts nachgereicht werden. Es macht daher Sinn, mit seiner benannten Stelle eine Zeitschiene zu besprechen, wann sind die Daten soweit ‚UDI-konform‘, wann das Qualitätsmanagement und so weiter.“
UDI-konform, MDR-konform: Keine Kleinigkeit
Ein Produkt „MDR-konform“ zu machen, ist alles andere als trivial, gibt Corinna Mutter von Spectaris zu bedenken: „UDI und Eudamed stellen für jedes Unternehmen ein vollständiges kleines IT-Projekt dar. Die Daten- und Systempflege muss kontinuierlich fortgeführt werden. Dies bedeutet eine hohe Kapazitätsbindung und ist vermutlich nicht ohne zusätzliches Personal und professionelle Hilfe im IT Bereich von außen zu schaffen.“
Investitionen, die von vielen kleinen und mittelständischen Unternehmen (KMU) schwer bis gar nicht zu stemmen sind. Darüber hinaus müssen sie ihre Daten aktuell und systemübergreifend konsistent halten und im Rahmen der Post-Market-Surveillance ebenfalls klinische Daten nach der Markteinführung sammeln, dokumentieren und auswerten. Einen solchen Mehraufwand schütteln die wenigsten Hersteller einfach so aus dem Ärmel.
Aufwand ist für KMU kaum zu stemmen
Die Folge: Laut einer Studie von Spectaris und dem Deutschen Industrie- und Handelskammertag wollen rund die Hälfte der 282 befragten Unternehmen ihre Produktlinien verringern. Etwa ein Drittel der Unternehmen, die ihre Erzeugnisse gemäß der MDR höher klassifizieren müssen, planen bereits, Produkte aus dem Programm zu nehmen. Das bestätigt auch Meinrad Kempf von Medical Mountains: „Allein personell ist es für KMU schwer, den zusätzlichen Verwaltungsaufwand zu stemmen. Wir treffen auf drei Reaktionen: Hersteller ziehen sich vom Markt zurück und werden zur ‚verlängerten Werkbank‘, Unternehmen werden verkauft oder aber ganz geschlossen. Eine fatale Entwicklung, da gerade die KMU mit neuen Ideen den Markt bereichern.“
Die Region um Tuttlingen ist besonders betroffen, da hier viele Hersteller von Produkten ansässig sind, die in die neugeschaffene Klasse Ir fallen: wiederverwertbare chirurgische Instrumente. Sie müssen sich nun von einer benannten Stelle zertifizieren lassen – von denen es wie gesagt im Oktober 2019 erst ganze fünf gab.
Medical Mountains bietet daher Workshops und Seminare zur MDR sowie Zertifikatslehrgänge an, um die Unternehmen zu unterstützen und Know-how im eigenen Betrieb aufzubauen. Auch das Land Baden-Württemberg stellt als erstes Bundesland speziell für seine mittelständische Medizintechnik-Branche Mittel für ein MDR-Soforthilfe-Programm bereit.
Basiswissen Unique Device Identification (UDI)
- Wer die UDI vergibt
Die eindeutige Produktidentifikation, auf englisch „Unique Device Identification“ (UDI) wird von verschiedenen Stellen zugeteilt. Zurzeit sind dies GS1, HIBCC und ICCBBA. Der Health Industry Barcode HIBC und der GS1-Code sind gleichermaßen als UDI-Code für Medizinprodukte geeignet. Der ICCBBA UDI-Code wird von der FDA – und damit voraussichtlich auch von der EU – zur Kennzeichnung von Produkten menschlichen Ursprungs (wie Blutplasma oder Transplantate) anerkannt. - Woraus die UDI besteht
Die UDI besteht aus zwei Teilen, der Produktkennung (UDI-DI) und der Produktionskennung (UDI-PI):
– der UDI-DI = UDI Device Identifier dient der Identifikation von Produkt und Hersteller und muss auf dem Produkt selbst und seiner Verpackung aufgebracht werden. In ihm sind Informationen enthalten wie zum Beispiel der Handelsname, Informationen über Wiederverwendbarkeit, Sterilität und so weiter – insgesamt etwa 20 Daten. Er enthält, je nach Zuteilungsstelle die GTIN (Global Trade Item Number) (GS1), die UPN (Universal Product Number) (HIBCC) oder den ISBT 128-PPIC (Processor Product Identification Code) (ICCBBA ).
– der UDI-PI = UDI Production Identifier markiert die Charge eines Produktes und damit unter anderem: Los- oder Chargennummern, Seriennummern, Verfalls- und/oder Herstelldaten.

Sonderrolle Basic UDI-DI
Eine Sonderrolle nimmt die Basic UDI-DI ein. Sie ist eine Art Modellkennzeichnung und umfasst gleichartige Produkte mit gleicher Zweckbestimmung und Risikoklasse, zum Beispiel mit unterschiedlichen Motorstärken. Sie dient als Schlüssel für die UDI-Datenbank, erscheint jedoch nicht auf dem Produkt oder dessen Verpackung. Und sie ist es auch, die von den Herstellern dann (voraussichtlich ab 26. Mai 2020) mit weiteren Datenelementen bei der UDI-Datenbank angegeben werden soll. Die Hersteller sind darüber hinaus verpflichtet, eine Liste aller vergebenen UDIs als Teil der technischen Dokumentation für jedes Medizinprodukt zu pflegen.
Wo muss die UDI erscheinen?
Ab 2021 müssen die ersten Medizinprodukte-Hersteller nicht nur das Produkt selbst, sondern auch jede übergeordnete Verpackung (mit Ausnahme der Container) mit der UDI kennzeichnen. Wichtig vor allem bei wiederverwertbaren Produkten: Der Code muss über die gesamte Nutzungsdauer lesbar bleiben – das ist zum Beispiel besonders für wiederverwendbare chirurgische Instrumente von Bedeutung, die viele Reinigungszyklen überstehen müssen.
Hier punktet das Lasermarkieren mit anschließender Passivierung – laut einer bereits 2017 von Foba Laser Marking + Engraving, Selmsdorf und der Add’n Solutions GmbH & Co. KG, Tuttlingen durchgeführte Studie: Die so aufgebrachte und geschützte UDI-Markierung blieb auf dem Edelstahl über mindestens 500 Sterilisations- und Reinigungszyklen klar lesbar.

Einige Hersteller, wie zum Beispiel Trumpf aus Ditzingen, bieten gleich Komplettpakete samt Software an. Da wird aus der Datenbank des Herstellers die korrekte UDI-Kennzeichnung erstellt, auf den Lasermarkierer geschickt und das Produkt mit UDI versehen – auf Wunsch mit anschließender Qualitätskontrolle und Dokumentation.
Ab wann UDI?
Die Kennzeichnungspflicht muss, abhängig von der Produktklasse, unterschiedlich schnell eingeführt werden:
- für Klasse III und Implantate: ab Mai 2021
- für Klasse IIa und IIb: ab Mai 2023
- für Klasse I: ab Mai 2025
Nützliche Informationen zu UDI und MDR
FAQ zur UDI:
https://ec.europa.eu/docsroom/documents/36664
Liste benannter Stellen nach MDR in Deutschland:
www.zlg.de/medizinprodukte/dokumente/stellenlaboratorien/benannte-stellen-eu-2017745-mdr/
Listen der benannten Stellen nach MDR in der EU:
https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=34
www.johner-institut.de/blog/regulatory-affairs/unique-device-identification-udi/
www.imdrf.org/documents/documents.asp
Leitfaden der EU zur UDI:
https://ec.europa.eu/growth/sectors/medical-devices/current-directives/guidance_en
https://ec.europa.eu/growth/sectors/medical-devices/new-regulations/guidance_en
NAKI – Nationaler Arbeitskreis zur Implementierung der neuen EU-Verordnungen über Medizinprodukte (MDR) und In-vitro-Diagnostika (IVDR)
https://www.bundesgesundheitsministerium.de/naki/?L=0
www.gesundheitsindustrie-bw.de/MDR-IVDR
Informationen zu UDI, die der Laserkennzeichnungsspezialist Foba zu Verfügung stellt
https://www.fobalaser.com/blog/de/faq-zu-udi-teil-2-wie-werden-udi-codes-sicher-gekennzeichnet/







 für Medizinprodukte? id=){kind=link}