
Die neue Medizinprodukteverordnung (Medical Device Regulation, MDR) 2017/745 beschreibt exakt, welche Voraussetzungen die Hersteller bei der technischen Dokumentation zu erfüllen haben. Dabei geht es im Anhang II um die technische Dokumentation im engeren Sinne und im Anhang III um die Dokumentation dessen, was der Hersteller tun will, um sein Produkt zu überwachen, sobald er es in Verkehr gebracht hat.
Wie aus der bisher geltenden MDD bekannt, dient die technische Dokumentation auch laut MDR dazu nachzuweisen, dass das Produkt konform zur Verordnung ist. Neu ist allerdings, dass sich die EU-Kommission vorbehält, die Anforderungen an die technische Dokumentation zu ändern, wenn der technische Fortschritt es erforderlich macht. Für die Hersteller bedeutet dies, dass ihre Dokumentation zum lebenden Dokument wird: Ihr Umfang und ihre Granularität können sich permanent ändern, und sie muss während des kompletten Lebenszyklus des Produktes aktualisiert oder ergänzt werden.
Änderungen möglich, Ressourcen erforderlich
Damit reagiert der Gesetzgeber darauf, dass Änderungszyklen von Normen und Gesetzen bisher sehr langwierig waren. Die EU-Kommission schafft sich die Möglichkeit, schneller zu reagieren. Ein Hersteller muss aber in jedem Fall Änderungen, die ohne oder nur mit sehr kurzer Übergangsfrist gültig wären, aktiv verfolgen und Ressourcen dafür vorsehen.
Die beiden MDR-Anhänge sind an vielen Stellen vergleichbar mit den Anforderungen aus der MDD. An einigen Stellen müssen aber Dinge ergänzt werden, manche Elemente kommen auch neu hinzu. Allerdings sind das oft alte Bekannte, da es um Inhalte geht, die in gängigen Normen gefordert werden. Hersteller, die sich an diese Normen halten, können damit auf Vorhandenes zurückgreifen.
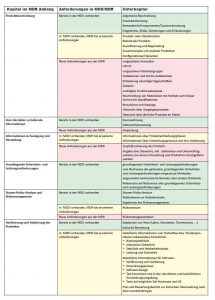
Ein Beispiel ist die Dokumentation der vorgesehenen Anwender. Die Norm für Gebrauchstauglichkeit, die IEC 62366, verlangt unter anderem das Erstellen und Dokumentieren der vorgesehenen Benutzerprofile. Im Rahmen des Usability Engineering werden diese in der Regel als Persona beschrieben, die eine bestimmte Benutzergruppe – wie den behandelnden Arzt – abdecken.
Ebenso verhält es sich mit dem Überblick über ähnliche, am Markt verfügbare Medizingeräte oder Vorgängerprodukte, den die DIN EN ISO 14971 fordert, um das Risiko zu minieren. Die Norm verweist auf den aktuellen Stand der Technik – und um diesen beurteilen zu können, braucht der Hersteller den Überblick über den Markt ohnehin und kann so die Vorgaben der MDR ohne Probleme erfüllen.
Erweitete Informationen zu Verifizierung und Validierung des Produktes
Im Vergleich zur MDD erweitert werden müssen unter anderem die Informationen zur Verifizierung und Validierung des Produktes. Ergebnisse der entsprechenden Tests mussten bisher schon nachgewiesen werden. Nun sollen auch detaillierte Informationen zum Testaufbau und Studienprotokolle geliefert werden, insbesondere zu Biokompatibilität, elektrischer Sicherheit, Stabilität und Haltbarkeitsdauer sowie Leistung und Sicherheit.
Wo Software im Spiel ist, werden detaillierte Informationen zur Verifizierung und Validierung gefordert sowie zum Entwicklungsprozess, dem Software-Design, den hausinternen Tests und den Tests in simulierten oder tatsächlichen Anwendungsumgebungen. Auch Tests auf möglicher Ziel-Hardware und OS müssen belegt werden.
Ganz neu ist das, was der Anhang III fordert: Das Beschreiben der Marktüberwachung nach dem Inverkehrbringen (Post Market Surveillance, PMS) gehört nun explizit zur technischen Dokumentation. Da diese die Konformität des Produktes zur Verordnung nach weisen soll, erscheint es konsequent, diesen Nachweis auch nach dem Inverkehrbringen zu aktualisieren. Denn sobald Probleme gemeldet werden, könnten diese die Risikoanalyse beeinflussen oder neue Risiko-Kontrollmaßnahmen erfordern.
Regelmäßig aktualisieren
Die technische Dokumentation regelmäßig zu aktualisieren, bringt je nach Art und Lebensdauer des Produktes einen erheblichen Aufwand mit sich. Der Aufwand ist hierbei allerdings abhängig von der Klassifizierung des Medizinprodukts.
- Für Medizinprodukte der Klasse I muss der Sicherheitsbericht erstellt, aber nur bei Bedarf aktualisiert werden.
- Für die Klassen IIa, IIb und III ist der Umfang des Berichtes größer (vgl. Artikel 86). Zudem gibt es eine festgelegte Zeitspanne, in der der Bericht aktualisiert werden muss. Bei Produkten der Klasse IIa ist dies bei Bedarf oder spätestens nach zwei Jahren der Fall. Für Produkte der Klasse IIb und III müssen Hersteller den Bericht bei Bedarf, aber mindestens jährlich aktualisieren.
Einen Plan zur Überwachung nach dem Inverkehrbringen gemäß Artikel 84 muss es jedoch immer geben – wobei der Inhalt im Anhang III beschrieben ist und damit die Anforderungen aus Artikel 83 erfüllt werden. Basierend auf diesem Plan muss der Hersteller einen Bericht über die Sicherheit erstellen.
Auch die Frage, wer für die technische Dokumentation verantwortlich ist, beantwortet die MDR: gemäß Artikel 15 ist es die „für die Einhaltung der Regulierungsvorschriften verantwortliche Person“, die die Unterlagen erstellt und aktualisiert.
Weiter Informationen zur Medical Device Regulation finden Sie auf unserer Themenseite







{kind=link}